ダブレットとは
シングルセル解析で現在主流になりつつあるのは10x Genomics Chromiumのようなドロップレットベースの方法である。この方法はドロップレットの中で磁気ビーズと細胞を封入し、mRNAの逆転写・増幅を行う (下図b)。ビーズにはポリT領域と、ビーズごとにユニークなバーコード配列がついたオリゴDNA (図c) が生えており、この上で細胞由来のmRNAが逆転写・増幅される仕組みである。この時にバーコードがついた状態でmRNAは逆転写・増幅されるので、配列解析時に、同じバーコードを持つリードは同一細胞に由来すると判別することができる。
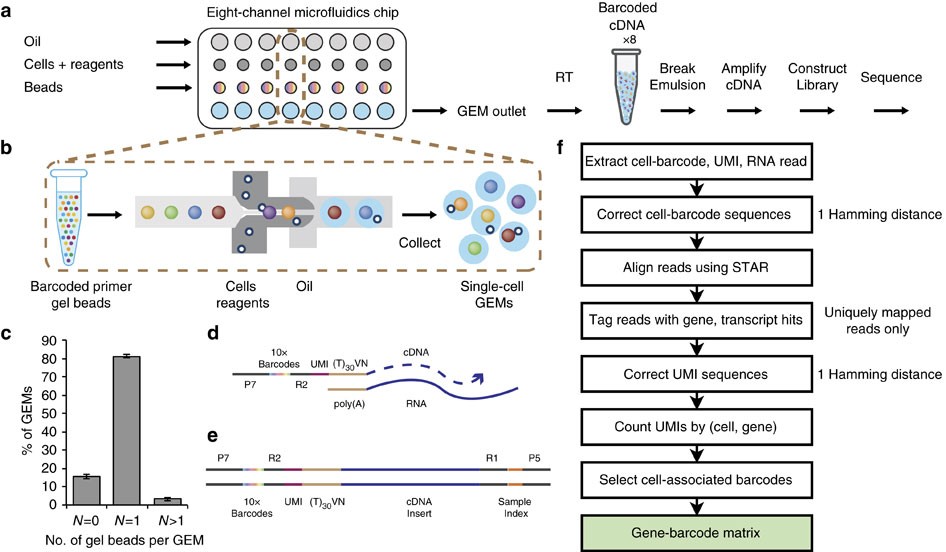
引用元:Zheng et al. “Massively parallel digital transcriptional profiling of single cells” Nat Commun. 2017 Jan 16;8:14049. doi: 10.1038/ncomms14049. CC-BY 4.0
一つのドロップレットの中にどのように一つの細胞を閉じ込めているのか?これは機械的に制御されているわけではない。ドロップレットの中に封入される細胞の数は確率的に決まり、ポアソン分布に従う。例えば、一つのドロップレットあたり平均0.5個の細胞が封入されるような濃度で実験を行えば、確率的に約60%, 30%, 8%のドロップレットには0個, 1個, 2個の細胞が封入される *1。すなわち細胞の濃度をある程度下げることで、ほとんどの細胞は単独でドロップレットの中に封入されることとなり、問題なくシングルセルレベルでの計測をすることができる、という算段である。
しかし実際には数%のドロップレットには複数個 (多くは2個) の細胞が入ってしまう (下図A)。この場合は、同じビーズ上でmRNAが処理され、同一のバーコードが付けられてしまう。したがって、配列解析時に異なる細胞由来のmRNAであることが区別できないため、複数個の細胞の遺伝子発現が、一つの細胞として、混ざった状態で検出されることになる。これがいわゆるダブレットと呼ばれるアーティファクトである。
ダブレットは解析結果を歪めかねない。一見するとユニークな細胞集団に見えて、実は単なるアーティフィシャルであることも多い (下図B)。

引用元:Wolock et al. “Scrublet: computational identification of cell doublets in single-cell transcriptomic data” bioRxiv. 2018 https://doi.org/10.1101/357368 CC-BY 4.0
Chromiumのマニュアル*2を見ると、1サンプルあたり10000細胞を解析する条件では、約7.6%がダブレットになるようだ。これは結構大きな影響がありそうなものだが、多くの論文ではあまりケアされていないように感じる。ダブレットを除くために、検出遺伝子数 (もしくはUMI数) が極端に多い細胞を解析から除外する前処理をよく目にするが、これは理想的とは言えない。検出遺伝子数は細胞種間で不均一であり、活性化した細胞や癌細胞は多くの遺伝子を発現しているからだ。
そんな中で2019年ごろから、よりエレガントにダブレットを解析から除外するための方法論が提案されてきた。ここではそれらの方法についてレビューする。
Scrublet, DoubletFinder
最初に報告された2つの手法 Scrublet*3とDoubletFinder*4。2019年4月の同時期にCell Systemsに公開されたこれらの手法はいずれも似たアプローチを提案している。基本的には以下の流れ。
- 入力: UMIカウント行列 (細胞 x 遺伝子)
- 1からランダムに細胞ペアをN組選出
- 2のペアのカウントデータをミックス 仮想ダブレットデータを作成
- 3を1と混ぜる
- Highly-variable geneの抽出 & PCAで次元圧縮 *5
- 主成分得点を使ってkNN graph *6 を描画
- 6の情報をもとにスコアを計算
Scrubletは以下の式でスコアが求まる*7。ダブレットはこのスコアが高くなる。
はユーザーが解析の際に指定する。(このパラメータが結果に影響することがgithubに記載されていた)
シングルセルデータにはダブレットとシングレットが混ざっているのでスコアの分布は二峰性を示す。この分布をもとにユーザーがカットオフをマニュアルで設定してダブレットを特定することが推奨されている。
DoubletDecon
2019年に報告された手法。Scrublet/DoubletFinderがなどのパラメータをユーザーが指定しなければならないことを問題視 *8。仮想ダブレットを作成するプロセスは共通しているが、発現変動遺伝子の情報を使って、過度に細胞を除きすぎないようにするプロセス (Rescue step) を設けている点が特徴。
- 入力: 正規化済み発現行列 (細胞 x 遺伝子)
- 前処理 (低発現遺伝子除外 & 質の低い細胞を除外)
- クラスタリング *9
- 3のクラスタごとに代表発現プロファイル (Centroid) を計算
- Cluster merging: 類似したクラスタをマージする *10
- 仮想ダブレットを作成: 5でできたクラスタのペアごとにランダムに細胞を選んで仮想ダブレットを作成
- 4をリファレンスに各細胞/仮想ダブレットの遺伝子発現プロファイルをDeconvolute。下図Eのような組成を計算する
- 7で得られた組成が仮想ダブレットの組成と類似している細胞は除外
- 8で除外した細胞を対象に3と同様の方法でクラスタリング
- 3と9の全クラスタ間で発現変動遺伝子を抽出
- Rescue step: 特異的遺伝子が検出されたクラスタは9からレスキューする

引用元:DePasquale et al. “DoubletDecon: Cell-State Aware Removal of Single-Cell RNA-Seq Doublets” bioRxiv. 2019 doi: https://doi.org/10.1101/364810 CC-BY 4.0
イントロではScrublet/DoubletFinderのパラメータ設定を問題視していたが、DoubletDeconも5, などのステップでad hocにユーザーがパラメータを決めなければならない様子。 Rescue stepは2つの細胞が混ざってできたダブレットであれば特異的に発現が動いている遺伝子はないという仮定に基づいている。この考え方は実装も楽だし、普遍的に使えそう。
SCDS
2020年に公開された手法。Co-expression based doublet scoring (cxds)とBinary classification based doublet scoring (bcds)という2つのスコアをもとにダブレットを推定する。
Co-expression based doublet scoring (cxds)
ある遺伝子が検出される細胞の割合を
とする。仮に2つの遺伝子
と
の発現が独立している場合は、2つのうちどちらかが検出される細胞の数
は次の二項分布に従う。これを利用すると2つの遺伝子の発現の排他性 (互いにExclusiveか) を検定することができる。
cxdsは以下のように求められる。通常は排他的に発現しているはずの遺伝子ペアが同時に発現しているほどスコアが高くなる仕組み。
Binary classification based doublet scoring (bxds)
- 入力: UMIカウント行列 (細胞 x 遺伝子)
- 1からM個のHighly-variable geneを選出 (default: M=500)
- 2からランダムに細胞ペアをN組選出
- 3のペアのカウントデータをミックス 仮想ダブレットデータを作成
- xgboostで仮想ダブレットと実際の細胞を分類する学習機を作る
- 仮想ダブレットへの分類確率をスコア (bxds) とする
まとめ
今回はダブレット推定をベースにした解析のアイデアがあったので世に出てきている手法を調べてみた*11。仮想ダブレットを作ってkNNやxgboost等で判別する手法が多い印象 *12 。性能や使いやすさについては追って検証したら追記する。
*2:https://kb.10xgenomics.com/hc/en-us/articles/360001378811-What-is-the-maximum-number-of-cells-that-can-be-profiled-
*3:https://www.sciencedirect.com/science/article/pii/S2405471218304745
*4:https://www.sciencedirect.com/science/article/pii/S2405471219300730
*5:Scrubletは1のみでPCA & その主成分へ3を後から射影
*6:k-nearest neighbor: 最近傍k個の細胞を結んだグラフ
*7:この式が導出された理論も論文に記載されていたがいまいち理解できず
*8:パブリックデータの場合はわからない場合も多い
*9:論文ではICGSを使用
*10:Batch effectなどで同一細胞が異なるクラスタとして検出される可能性を考慮しているらしい
*12:DNNで学習する手法 Soloも最近提案されてた https://www.sciencedirect.com/science/article/pii/S2405471220301952