Context-specific metabolic modeling
Context-specific metabolic modelingとは、トランスクリプトーム・プロテオーム等の発現情報を用いて、Recon3DやHuman1等の全代謝反応を含むGenome-scale metabolic model (GEMs)から、解析対象 (e.g. 特定の細胞、組織) で機能している・していない代謝反応のみを含むGEMsを抽出する手法。
2008年頃から色々な手法が提案されており、特に新しい研究分野ではないのだが、個人的なプロジェクトで似たような方法を試してみたいと考えている。ほとんどがMATLABで実装されていること (筆者はオープンソースで実装したい)、手法に応じて結果が大きく異なることが報告されていること *1 など、手法を理解していない状態で試すと危ない雰囲気なので、簡単にまとめてみようと思う。
Context-specific metabolic modelingの手法は以下のレビュー論文では3つのグループに分類されている。ここでは各グループの代表的な手法について調べる。
GIMME-like family
上記論文では「発現量の低い遺伝子・タンパク質に対応する代謝反応の流束を最小化する」制約条件を利用した手法がGIMME-likeと分類されている。
GIMME
GIMMEは2008年にPalssonらによって発表された最初期の手法。(i) 遺伝子発現 (or タンパク発現) データ、(ii) 対象生物種のGEM、(iii) 発現の有無を決めるための閾値 (e.g. この閾値を上回る遺伝子は「発現あり」、下回る場合は「発現なし」とみなされる)、(iv) Required Metabolic Functionalities (RMF) (生物学的な知識に基づき存在していると考えられる代謝機能) を入力として受け取る。原著論文では「培地中の炭素源が生育につながる*2」ことをRMFとして設定している。
以下のステップで組織・細胞特異的なGEMsを構築する。
- 発現の無い (閾値を下回る) 遺伝子・タンパク質に対応する代謝反応をGEMから除外する
- RMFを実現できるように除外した代謝反応の一部を元に戻す
- RMFを最大化する代謝流束をFBAで推定する
- RMFがある値以上 (論文では3.で求めた最大値の0.9倍以上を基準としている) となる範囲で以下のInconsistency Score (IS) が最小となる代謝流束をFBAで計算する
Inconsistency scoreは下図のように設定されている。赤い矢印は対応する遺伝子の発現量が閾値 *3 を下回っている代謝反応を表す。最適な閾値を決める方法は本論文では特に提案されていない。閾値をいくつか試して結果への影響が小さかったことを確認している、とのこと。*4 上記のようにGIMME-like familyは遺伝子・タンパク質発現とある程度齟齬のない代謝流束で、Biomass functionなどの目的関数を最大化することを目指す手法である。
: Inconsistency Score
: 反応
の流束
: 閾値
: 反応
引用元:Becker SA, Palsson BO. Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol. 2008 May 16;4(5):e1000082. doi: 10.1371/journal.pcbi.1000082. PMID: 18483554; PMCID: PMC2366062.
iMAT-like family
上記レビュー論文では「低発現遺伝子に対応する代謝反応をできるだけ削り、高発現遺伝子に対応する代謝反応をできるだけ残す」最適化を行う手法をiMAT-likeと呼んでいた。GIMME-like familyは遺伝子発現データとの齟齬の少ない制約条件下でBiomass functionなどのRMFを最大化する代謝状態を探索する方法であったが、iMAT-like familyの手法はRMFを考慮せず、シンプルに遺伝子発現データとの齟齬の少ない代謝状態を探索する。
iMAT
iMATは2008年にShlomi・Palsson・Ruppinらによって提案された手法。(i) 遺伝子発現 (or タンパク発現) データ、(ii) 対象生物種のGEM、(iii) 発現レベルをHigh/Normal/Lowに分けるための閾値、(iv)パラメータ (後述)を入力として受け取る。
以下の制約条件下で目的関数が最大となる代謝流束を見つける。この目的関数を最大化することで、高発現遺伝子に対応する代謝の流束ができるだけ非ゼロの値になり、低発現遺伝子に対応する代謝の流束がゼロになるような、遺伝子発現データとの齟齬の少ない代謝状態を探索できる。下記論文のFig. 1がイメージをつかみやすい*5。この問題はパラメータが整数値なので混合整数線形計画法 (MILP)で最適化を行う。MILPは線形計画法(LP)よりも最適解の探索が困難であるため*6、おそらくGIMMEなどのLPベースの手法よりも計算時間を要すると思われる。
- 目的関数:
- 制約条件
INIT (tINIT)
INITはJens Nielsenらのグループが2012年に発表した手法。その後2014年に改良版のtINITが発表された。INITは以下のように定式化されている。原著論文を読んでいてもわからない点があったのでレビューを参考にした。iMATと同様に一部のパラメータが整数値なのでMILPで最適化を行う。
- 目的関数:
- 制約条件
引用元:Agren R, Bordel S et al. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput Biol. 2012;8(5):e1002518. doi: 10.1371/journal.pcbi.1002518. Epub 2012 May 17. PMID: 22615553; PMCID: PMC3355067.
https://www.embopress.org/doi/full/10.1002/msb.145122www.embopress.org
MBA-like family
遺伝子発現データを元にGEMから発現の無い枝を取り除く(Pruning)する手法。この手法はGIMME-like familyやiMAT-like familyのように特定の代謝状態を探索するのではなく、
MBA
MBAは2010年にJerby・Shlomi・Ruppinによって提案された手法。下記の目的関数を最大化するPartial model (PM)を以下のアルゴリズムで探索する*12。Globam model (GM) から少しずつ不必要な反応を削っていくアプローチである。2. でCore reactions () に含まれない反応の集合
を抽出。3.以降のFor loopでは、
に含まれる反応
を一つずつKOして、その結果Inactiveになる反応を除外する (枝葉を切る, pruning)。
CheckModelConsistencyはモデルから反応
を除外したときにInactiveになる反応を返す関数。
2.においてに含まれる反応
の順番はランダムに決める。反応
の並びによって最終的なGEMの形が変わるので、論文ではpruningを1000回行い、できた1000個のGEMから共通性の高い領域を抽出している。
- 目的関数:
アルゴリズム
 引用元:Jerby, Livnat et al. “Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism.” Molecular systems biology vol. 6 (2010): 401. doi:10.1038/msb.2010.56
引用元:Jerby, Livnat et al. “Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism.” Molecular systems biology vol. 6 (2010): 401. doi:10.1038/msb.2010.56
mCADRE
mCADREはMBAを改良した方法で2012年に発表された。MBAがFor loopのiterationの順番をランダムに決めていたことを問題視。近傍にCore reactionがあるかを考慮したスコアを設定し、Non-core reaction に含まれる反応を並べることで、一意な結果が得られる(かつMBA論文のように1000回 iterationを実施する必要がないのでより短い時間で計算できる)手法を提案。
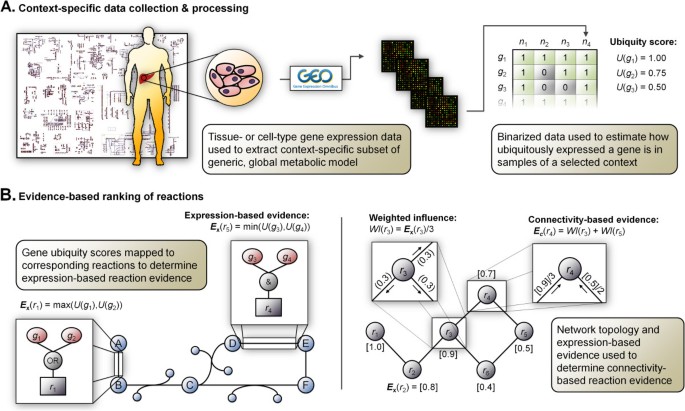
引用元:Wang, Y., Eddy, J.A. & Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst Biol 6, 153 (2012). https://doi.org/10.1186/1752-0509-6-153
Pythonで上記の手法を試すには
Pythonでは以下のライブラリに各手法が実装されている。Context-specific modelingの関数・クラスについての説明がほとんど存在しないツールもあるので、本当に使えるのか?だが、実装する際に参考になると思われる。
*1:https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006867
*2:Biomass産生につながる
*3:この論文の場合は12.0
*4:Data not shownだが
*6:https://www.msi.co.jp/nuopt/introduction/algorithm/mip.html
*7:一般的なFVAの制約条件:定常状態
*8:これも一般的なFVAの制約条件:事前情報に基づく各代謝の最大・最小流束
*9:Tissue jにおける発現値を全Tissueの平均値と比較
*11:これによってこの物質の産生に関わる代謝反応が除外されることを防ぐ
*12:なぜかこの論文では差集合をバックスラッシュでなくスラッシュで表しているので注意