はじめに
10X ChromiumをはじめとするDroplet-basedのシングルセル解析を行う場合にAmbient RNA contaminationが問題になることがある。Ambient RNAは細胞に由来しないRNAであり、処理中に壊れた細胞から溶出したRNAや、コンタミネーションなどが由来となる。Ambient RNAはDropletに混入して、あたかも細胞に由来するRNAのように検出されるため、遺伝子発現データを歪める恐れがある。ここではAmbient RNAの影響をデータから除外するための方法についてまとめる。
SoupX
SoupXは2020年にGigaScienceで発表された手法。以下のステップでAmbient RNAの影響を取り除く。
- Empty dropletの情報を使ってAmbient RNAのプロファイルを推定する
- 細胞種特異的なExclusive marker geneset
を同定
- Ambient RNAの混入率
を細胞ごとに推定する
- Exclusive marker geneの発現情報をもとに推定する
- Exclusive marker geneの発現情報をもとに推定する
- 各細胞の発現プロファイルを補正する

引用元:Young et al. “ SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data” GigaScience. 2020. https://doi.org/10.1093/gigascience/giaa151. CC-BY 4.0
https://academic.oup.com/gigascience/article/9/12/giaa151/6049831academic.oup.com
DecontX
DecontXは2020年にGenome Biologyで発表された手法。トピックモデリングのような以下の階層ベイズモデルを使ってAmbient RNAを分離する。入力データはUMIカウントデータで、細胞のクラスタリングを事前情報として与える必要がある。
: 細胞種 (細胞クラスタ) の数
: 細胞数
: 細胞jのUMI数
: 細胞jのUMI tが細胞に由来する確率
: 細胞jのUMI tが細胞種
に由来するとき、遺伝子gに由来する確率
: 細胞jのUMI tがAmbient RNAに由来する確率
: 細胞jのUMI tが細胞種
: Ambient RNAに含まれる各遺伝子の割合
: パラメータ
の事前分布
: パラメータ
の事前分布
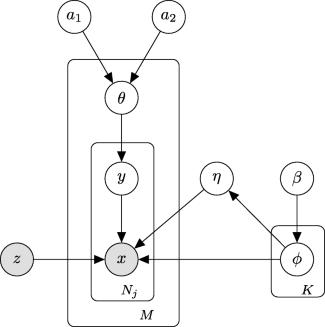
引用元:Yang et al. “Decontamination of ambient RNA in single-cell RNA-seq with DecontX” Genome Biol. 2020. https://doi.org/10.1186/s13059-020-1950-6. CC-BY 4.0
genomebiology.biomedcentral.com
CellBender remove-background
2019年にbioRxivに公開された方法。以下のようにUMIカウントデータをモデリングしてバックグラウンドの推定を行う。この手法ではバックグラウンドがAmbient RNAに由来するだけでなく、Barcode swapping (PCRでキメラ配列が形成されることで誤ったバーコードが付与される) についても考慮している点が特徴である。
: Droplet nにおけるノイズ除去後の遺伝子gの割合 (NN=Neural network)
: Droplet nを表す潜在ベクトル (N(0, 1)の正規分布を事前分布に設定)
: Size factor (=バックグラウンド由来のUMI量)
]: Size factor (=細胞由来のUMI量)
: ハイパーパラメータ *5
: Droplet nに細胞が含まれていたか否か
: 予想解析細胞数をもとに決める
: Droplet nにおける他の細胞由来 (上述のBarcode swappingによる) UMIの割合
: 細胞nにおける遺伝子gのUMI数
: 全細胞での遺伝子gの平均割合 (Barcode swappingでは他の全細胞からランダムに転写物が混入するので)
: Ambient RNAにおける遺伝子gの割合 (これは推定する)

引用元:Fleming et al. “CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets” bioRxiv. 2019. https://doi.org/10.1101/791699 . CC-BY 4.0