COBRApy
以前の記事で触れたFlux balance analysis (FBA) にはMATLAB用のパッケージCOBRAが使われている論文が多い。筆者の解析環境にはMATLABはインストールされていないので*1、COBRAの機能をPythonで使えるようにしたパッケージCOBRApyを使おうと思うが、COBRAと比較してチュートリアルが充実しておらず*2、実際に使う上で少しハードルが高い印象を受けた。この記事では、COBRAの開発グループが2017年にNature Biotechnologyへ掲載した論文のデータをCOBRApyを使って再現してみることで、COBRApyの機能を理解しようと思う。この論文は、ゲノムデータ・文献情報をもとに腸内細菌773種のGEMs (Genome-scale metabolic models)のセットAGORAを作成し、各細菌の代謝の特徴や、2種の細菌が共存する場合の代謝相互作用をFBAを使って推定している。
本記事の検証に使ったコードはgithubにアップした。
データを準備
- AGORA GEMs: https://github.com/VirtualMetabolicHuman/AGORA
- 細菌のメタデータ: Supplementary Table 5
- 代謝・代謝物のメタデータ: Supplementary Table 14
- 食事成分データ: Supplementary Table 15に記載。ここではtsvを用意した。
- 培地データ: Supplementary Table 13に記載。ここではtsvを用意した。
git clone https://github.com/tmaruy/cobrapy.git cd cobrapy # AGORAをダウンロード git clone https://github.com/VirtualMetabolicHuman/AGORA.git # メタデータをダウンロード curl "https://static-content.springer.com/esm/art%3A10.1038%2Fnbt.3703/MediaObjects/41587_2017_BFnbt3703_MOESM6_ESM.xlsx" --output Magnusdottir-2017-Nat_Biotechnol/41587_2017_BFnbt3703_MOESM6_ESM.xlsx curl "https://static-content.springer.com/esm/art%3A10.1038%2Fnbt.3703/MediaObjects/41587_2017_BFnbt3703_MOESM10_ESM.xlsx" --output Magnusdottir-2017-Nat_Biotechnol/41587_2017_BFnbt3703_MOESM10_ESM.xlsx
Fig. 1
Fig. 1bではAGORAに存在する各細菌のGEMsの特性 (e.g. GEMsに存在する代謝反応・代謝物の数) を示している。これを再現してみる。AGORAはGEMsをSBML形式 *3 で保存している。COBRApyではこれをcobra.Model型のインスタンスとして読み込む。cobra.Model型の基本的な特性やモデルの読み込みについてはこのノートやCOBRApyのマニュアルを参照されたい。
import numpy as np import pandas as pd import glob import cobra from plotnine import * # ggplot2-like plotting in python # GEMのリストを取得 sbml_files = np.sort(glob.glob("AGORA/CurrentVersion/AGORA_1_03/AGORA_1_03_sbml/*xml")) bacteria_names = [f.split("/")[-1].replace(".xml", "") for f in sbml_files] # 細菌メタデータ s_meta = pd.read_excel("Magnusdottir-2017-Nat_Biotechnol/41587_2017_BFnbt3703_MOESM6_ESM.xlsx", skiprows=2) s_meta = s_meta.loc[s_meta.ModelAGORA.isin(bacteria_names), :] # GEMが見つからない細菌を除外 # GEMから代謝反応、代謝物質のリストを抽出 s2r = []; s2m = [] for s in tqdm.tqdm(s_meta.ModelAGORA): file = "AGORA/CurrentVersion/AGORA_1_03/AGORA_1_03_sbml/{s}.xml".format(s=s) model = cobra.io.read_sbml_model(file) s2r.append(pd.DataFrame({"species": s, "reactions":[x.id for x in model.reactions]})) s2m.append(pd.DataFrame({"species": s, "metabolites":[x.id for x in model.metabolites]})) s2r = pd.concat(s2r, ignore_index=True) s2m = pd.concat(s2m, ignore_index=True) # Fig. 1b: 代謝反応の数 figmat = s2r.groupby("species").size().rename("n_reaction").reset_index() figmat = pd.merge(figmat, s_meta.rename(columns={"ModelAGORA":"species"}).loc[:, ["species", "Phylum"]], how="left", on="species") ( ggplot(figmat) + geom_boxplot(aes(x="Phylum", y="n_reaction")) + theme_bw() + theme(axis_text_x=element_text(angle=90), figure_size=(5,3)) + scale_y_continuous(limits=[500, 2000], breaks=[500, 1000, 1500, 2000]) + ggtitle("Number of reactions") ) # Fig. 1b: 代謝物質の数 figmat = s2m.assign(metabolite_id = lambda x: [y[:-3] for y in x.metabolites], location = lambda x: [y[-2] for y in x.metabolites]).\ # merge extra/intracellular metabolites groupby(["species"]).metabolite_id.nunique() figmat = pd.merge(figmat, s_meta.rename(columns={"ModelAGORA":"species"}).loc[:, ["species", "Phylum"]], how="left", on="species") ( ggplot(figmat) + geom_boxplot(aes(x="Phylum", y="metabolite_id")) + theme_bw() + theme(axis_text_x=element_text(angle=90), figure_size=(5,3)) + scale_y_continuous(limits=[400, 1300], breaks=[400,600,800,1000,1200]) + ggtitle("Number of metabolites") )
結果
Fig. 1bのデータと類似した傾向が認められた。


Supplementary Table 6
FBAでは、培地条件など、周辺環境中の物質の組成を変えることによる影響をシミュレーションすることができる。Supplementary Table 6では、2種類の食事 (Western dietとHigh fiber diet) を摂取した場合、酸素存在化・無酸素条件下の最大生育率 *4 を細菌ごとに計算している。環境中成分の設定方法は本家のマニュアルに記載されている。有酸素条件では、このページを参考に、細菌による酸素取り込みの流束を10 mmol/gDW/h *5 とした。
ここでは、以下のようなコードでSupplementary Table 6のデータを再現してみた。全773種について計算すると時間がかかるので、ランダムに選んだ50種について、4種類の生育条件 (2種類の食事と2種類の酸素条件) での生育率を推定し、論文のデータと比較した。
growth_rate_check = {"species":[], "Western_Anaerobic":[], "Western_Aerobic":[], "HFD_Anaerobic":[], "HFD_Aerobic":[]}
for s in tqdm.tqdm(species):
growth_rate_check["species"].append(s)
file = "AGORA/CurrentVersion/AGORA_1_03/AGORA_1_03_sbml/{s}.xml".format(s=s)
model = cobra.io.read_sbml_model(file)
# Western diet
with model:
# Anaerobic
medium = model.medium
for k in medium.keys(): medium[k] = 0
for ex,val in zip(diet["Exchange reaction ID"], diet["Western Diet"]):
if model.reactions.has_id(ex): medium[ex] = val
model.medium = medium
op = model.optimize()
growth_rate_check["Western_Anaerobic"].append(op.objective_value)
# Aerobic
medium["EX_o2(e)"] = 10
model.medium = medium
op = model.optimize()
growth_rate_check["Western_Aerobic"].append(op.objective_value)
# High fiber diet
with model:
# Anaerobic
medium = model.medium
for k in medium.keys(): medium[k] = 0
for ex,val in zip(diet["Exchange reaction ID"], diet["High fiber Diet"]):
if model.reactions.has_id(ex): medium[ex] = val
model.medium = medium
op = model.optimize()
growth_rate_check["HFD_Anaerobic"].append(op.objective_value)
# Aerobic
medium["EX_o2(e)"] = 10
model.medium = medium
op = model.optimize()
growth_rate_check["HFD_Aerobic"].append(op.objective_value)
growth_rate_check = pd.DataFrame(growth_rate_check)
結果
論文と使っているAGORAのバージョンが違うからか、最適化に使っているソルバーの違いからか、若干の差異はあるが、論文のデータとおおよそ相関する結果が再現できた。

Fig. 3
ここでは各細菌がどの物質を利用して生存できるか、どの物質を産生できるか、をFVAで解析している。FVAは目的関数がある条件を満たす場合に各代謝反応が取りうる流束の範囲を推定する方法である。詳しい解説は次の論文を参照のこと。
bmcbioinformatics.biomedcentral.com
論文によると生育率が0.001 を上回る状況(細菌が増加する状況)で各代謝がとりうる流束の範囲をFVAで推定しているらしい。ここでは
fraction_of_optimumというオプションを使って同様の条件を設定してみた。
論文と同様に、Exchange reaction *6 の最小流束が0よりも下回る場合に「細菌がその物質を環境から取り込む」、最大流束が0よりも高い場合に「細菌がその物質を環境へ分泌する」と考える。
fig3_reactions = pd.read_table("Magnusdottir-2017-Nat_Biotechnol/fig3_metabolites.tsv") # fig.3に含まれている反応のリスト def fig3_metabolic_potential(species): species2potential = [] for s in tqdm.tqdm(species): file = "AGORA/CurrentVersion/AGORA_1_03/AGORA_1_03_sbml/{s}.xml".format(s=s) model = cobra.io.read_sbml_model(file) op = model.optimize() value = op.objective_value fva = cobra.flux_analysis.flux_variability_analysis(model, fraction_of_optimum=0.001/value) fva_ex = fva.loc[[i.startswith("EX") for i in fva.index],:].\ reset_index().rename(columns={"index":"Reaction"}).\ merge(fig3_reactions, how="right", on="Reaction").\ loc[:, ["Reaction", "Reaction name", "Category", "minimum", "maximum"]].\ assign(species = s) species2potential.append(fva_ex) return pd.concat(species2potential)
結果
主要なPhylumであるActinobacteria, Bacteroidetes, Firmicutes, Proteobacteriaの代謝能を比較してみた。ここでは20種ずつ計算した。Glycerolの代謝がBacteroidetesで少なく、Succinateの代謝がFirmicutesで少ないなど、Fig3で確認できた傾向が再現できた。
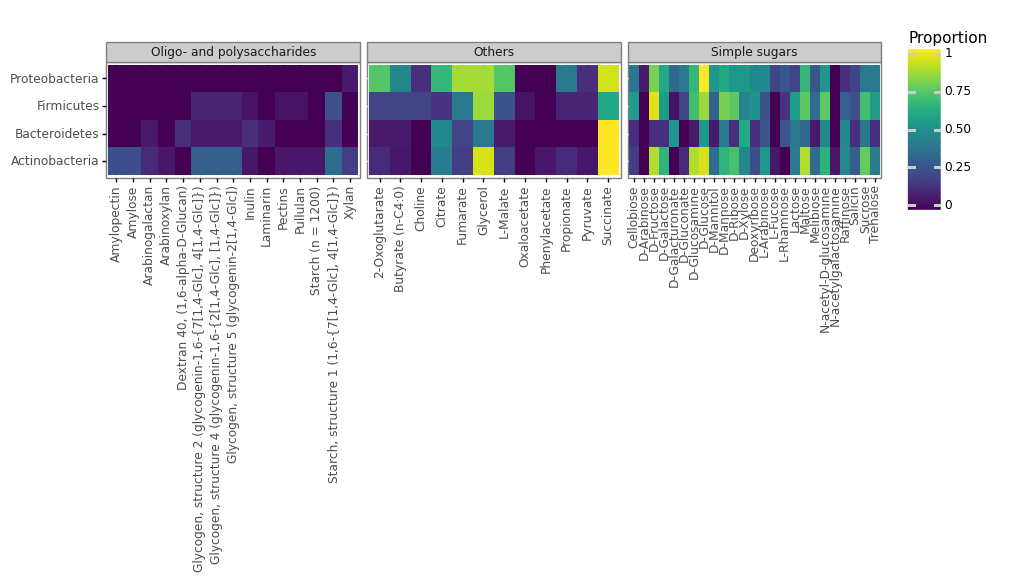
またFusobacterium 16種の代謝能を調べたところ、論文で触れられているのと同様に、全種がButyrate産生のポテンシャルを有していることがFVAで推測された。
Fig. 4
Fig. 4ではBacteroides caccae ATCC 43185とLactobacillus rhamnosus GGの生育条件を調べている。ここではDMEM 6429という培地に特定の物質を加えることで生育が変わるかどうかを推定している。
dmem = pd.read_table("Magnusdottir-2017-Nat_Biotechnol/Medium-DMEM_6429.tsv") s = "Bacteroides_caccae_ATCC_43185" file = "AGORA/CurrentVersion/AGORA_1_03/AGORA_1_03_sbml/{s}.xml".format(s=s) model = cobra.io.read_sbml_model(file) medium = model.medium for k in medium.keys(): medium[k] = 0 for ex,val in zip(dmem["Exchange reaction ID"], dmem["Uptake rate"]): if model.reactions.has_id(ex): medium[ex] = val model.medium = medium op = model.optimize() print("DMEM with Vitamin K, Hemin, Arabinogalactan: {g}".format(g=op.objective_value)) medium["EX_arabinogal(e)"] = 0 model.medium = medium op = model.optimize() print("DMEM with Vitamin K, Hemin: {g}".format(g=op.objective_value)) #medium["EX_mqn7(e)"] = 0 #medium["EX_mqn8(e)"] = 0 medium["EX_pheme(e)"] = 0 model.medium = medium op = model.optimize() print("DMEM only: {g}".format(g=op.objective_value))
結果
論文での予測と同様にBacteroides caccae ATCC 43185はArabinogalactan存在化で生育率が上昇することが示唆された。Lactobacillus rhamnosus GGも論文と同様にNicotinic acidが生育に寄与することが示唆された。
- Bacteroides caccae ATCC 43185
- DMEM with Vitamin K, Hemin, Arabinogalactan: 0.5330796480552902
- DMEM with Vitamin K, Hemin: 0.2768354439324188
- DMEM only: 0.2768354439324188
- Lactobacillus rhamnosus GG
- DMEM with Vitamin K, Hemin, Arabinogalactan: 0.0
- DMEM with Vitamin K, Hemin, Arabinogalactan, Nicotinic acid: 0.312092469574153
- DMEM with Nicotinic acid: 0.312092469574153
- DMEM with Nicotinic acid w/o L-alanine: 1.4612128398466525e-16
Fig. 4dで行われているような細菌間相互作用の解析も興味深いので次の記事で触れたい。