bioBakery
bioBakeryはHarvardのCurtis Huttenhowerのグループが開発したマイクロバイオーム解析のツール群である。マイクロバイオーム関連の研究を見ているとよく名前が出てくるツールが多数含まれているので、改めてどのようなツールが提供されているのか、気になったものについて調べてみる。
- MetaPhlAn: ショットガンメタゲノムデータから由来細菌種・菌叢組成を推定
- HUMAnN: ショットガンメタゲノムデータからを機能遺伝子を同定・機能プロファイルの推定
- PICRUSt: メタ16Sデータから菌叢の機能プロファイルを予測
- MelonnPan: ショットガンメタゲノムデータから機械学習で代謝物プロファイルを予測
- StrainPhlAn: マーカー遺伝子中の多型情報をもとにStrainレベルの情報を取得
- WAAFLE: メタゲノムアッセンブリで得られたコンティグから遺伝子水平伝播を検出
- KneadData: ショットガンメタゲノムデータの前処理ツール
MetaPhlAn3
MetaPhlAnは、約17,000種(~13,500 bacterial and archaeal, ~3,500 viral, and ~110 eukaryotic)のゲノムから構築した、系統特異的な遺伝子のリストをメタゲノムデータから探索することで、細菌叢の組成を推定する方法である。細菌の全ゲノムデータへマッピングするよりもはるかに効率よく菌叢組成を推定できる。2012年に公開された最初のバージョンから徐々に遺伝子リストが更新されており、2020年にバージョン3が発表された。
# データベース構築 metaphlan --install --bowtie2db ${db} # $dbにデータベースのファイルが作られる # 細菌種同定 metaphlan \ ${fastq1},${fastq2} \ # Input: ペアエンドの場合はカンマで区切って指定する --bowtie2db ${db} \ # DB --index latest \# DBのバージョン (default: "latest" 最新のDBを使う $dbになければインストールする) --input_type fastq \ # --nproc 8 \ # CPU数 --tax_lev {a, k, p, c, o, f, g, s} \ # 同定する系統レベル (defalt: a (all)) --unknown_estimation \ # 未知の細菌がどの程度いるかを推定する --bt2_ps {very-sensitive, sensitive, very-sensitive-local, sensitive-local} \ # bowtie2のモード (default: very-sensitive) --min_mapq_val 5 \ # defualt: 5 --add_viruses \ # ウイルスも検出する --ignore_eukaryotes \ # 真核生物を考慮しない --ignore_bacteria \ # バクテリアを考慮しない --ignore_archaea \ # アーキアを考慮しない --read_min_len 70 \ # この長さより短いリードは考慮しない -bowtie2out ${bowtie2_out} \ # 中間ファイル (bowtie2の出力) これを残しておくと再計算がすぐできる -s ${sam} \ # -o ${result} \ # -t rel_ab # return relative abundance (default: rel_ab)
Kraken/Bracken
Kraken/BrackenはbioBakeryではないがこちらもよく目にするのでチェック。MetaPhlAnと同様にショットガンメタゲノムデータの細菌種同定・菌叢組成推定のためのツール。Krakenは系統特異的なk-merの情報を元に各リードの由来生物種をLowest Common Ancestor *1で推定する。BrackenはKrakenの後に開発された手法。Lowest Common Ancestorでは多くのリードが種・属レベルでアサインすることが困難なので、ベイズ的手法を使ってどの種にアサインされるか確率的な推定を行う。
は以下のように求める。ゲノム
からリードと同じ長さ
の配列を生成し、そのうちKrakenで
にアサインされる割合を調べる。
:
から生成した長さ
の配列のうち
にアサインされた数
: 長さ
のゲノム
は以下のように求める。ゲノム
にアサインされたリードの数から推定する。
以上のように求めたを用いて、
にアサインされたリードの数から、各ゲノム
にアサインされるリード数を推定する。
# krakan2 kraken2 \ --db ${database} \# k-mer database --confidence 0.1 \ # threshold (default: 0.0) --threads 8 \ #スレッド数 --output ${kraken_output} \# Output file 1 (各リードがどの系統にアサインされたか) --report ${kraken_report} \# Output file 2 (各系統にLCAでアサインされたリードの数) --gzip-compressed \# Inputがgzipされている場合は指定する必要あり --memory-mapping \# k-mer databaseをメモリにロードしたくない場合 --paired ${forward_reads} ${reverse_reads} # bracken bracken \ -d ${database} \# k-mer db (same as kraken) -i ${kraken_report} \# Input from kraken -w ${bracken_output1} \# Output file 1 (Brackenのアルゴリズムで推定された各系統のリード数) -o ${bracken_output2} \# ${bracken_output1}の内容をKrakenのフォーマットで出力したもの -r 100 \# リード長 (上記の計算で使用する) -l {S, G, F} # S: species, G: genus
HUMAnN
HUMAnNは機能遺伝子の同定・菌叢の機能プロファイリングを効率的に行うパイプライン。(i) Prescreen: MetaPhlAnで細菌種特異的なマーカー遺伝子を探索 (ii) Nucleotide search: 同定された細菌種のゲノムをリファレンスとしてIndexを作成し、マッピング (iii) Translated search: マップされなかったリードをUniref90に対してDiamondでblastx-likeな検索を実施、という3ステップ。
# ペアエンドのデータを連結 (humannはペアの情報を考慮しない) cat ${forward_fastq} ${reverse_fastq} > ${fastq} # humann \ -i ${fastq} \# Input file -o ${output_directory} \# 結果が出力されるディレクトリ --taxonomic-profile taxonomic_profile.tsv \# --remove-temp-output \# 中間ファイルを消す (default: off) --o-log ${log_file} \# ログファイル (default: temp/sample.log) --output-basename ${basename} \# 出力ファイルのPrefix --output-format {tsv, biom} \# default: tsv --bypass-prescreen \# Prescreenをスキップ (Nucleotide searchには全細菌のゲノムを使う) --bypass-nucleotide-search \# Nucleotide searchをスキップ --bypass-translated-search \# Translated searchをスキップ --threads 8 \# スレッド数 --nucleotide-database ${database}
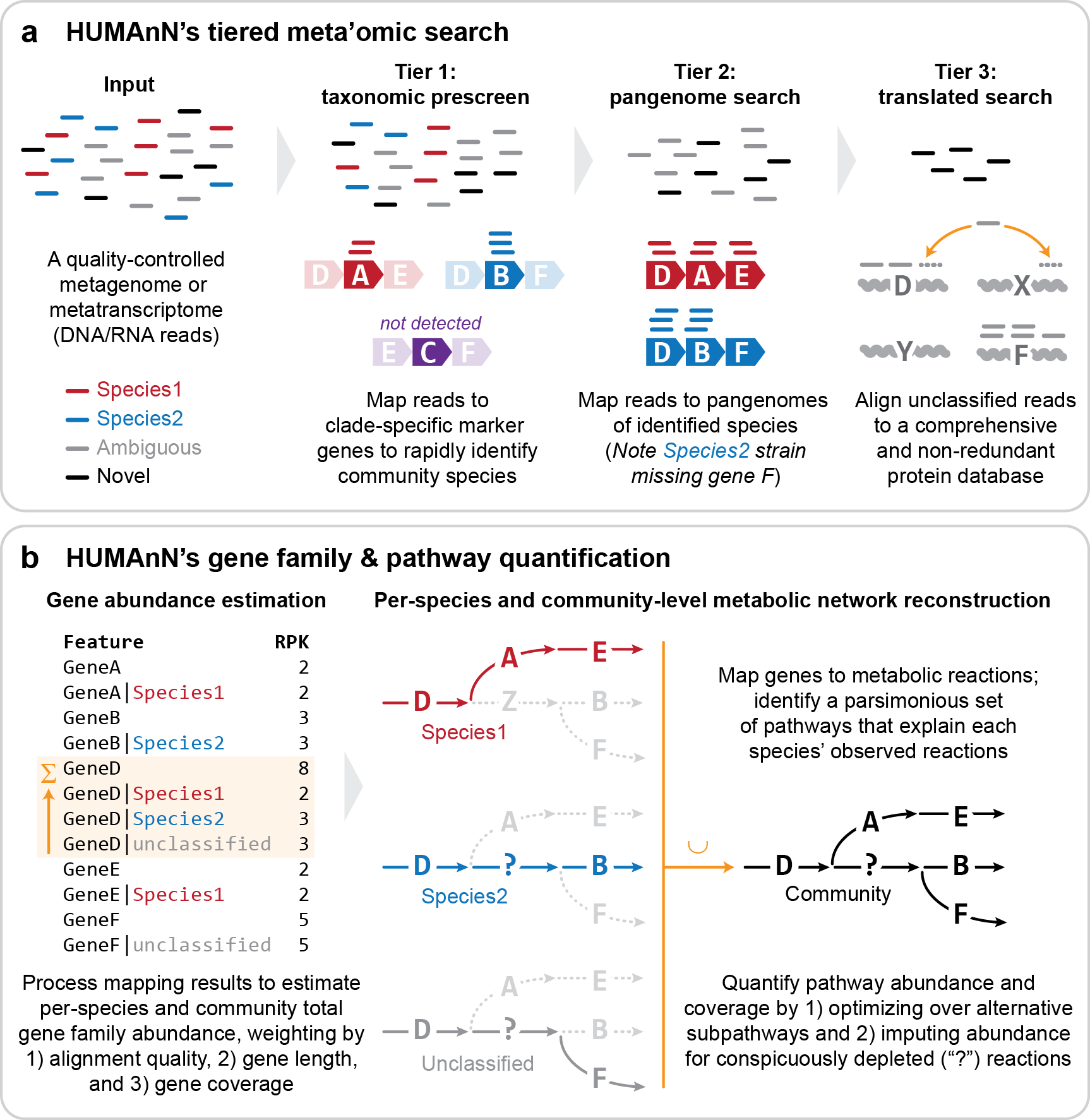
引用元:https://github.com/biobakery/biobakery/wiki/humann3
KneadData
ショットガンメタゲノムデータの前処理のためのツール。(i)低クオリティリードの除外と、(ii)アダプター配列の除去をTrimmomaticで、(iii)リピート領域に由来するリードの除去をTRFで、(iv)ホストゲノムに由来するリードをBowtie2で除外する。ホストゲノムのリファレンスにはhg37*3に加えて、Decoy Genomeと呼ばれるヒトゲノム由来ながらhg38にマッピングされないリードから構築されたコンティグ*4や、公共バクテリアゲノムによくコンタミしていることが報告されているヒト由来配列*5を用いる。メタトランスクリプトームデータの場合はリファレンスにはホストのトランスクリプト配列を用いる。リファレンスはコマンドkneaddata_databaseで既に構築されているものをダウンロードすることができる。
# ゲノムの場合 ($REFDIRにダウンロードされる) kneaddata_database --download human_genome bowtie2 $REFDIR # トランスクリプトの場合 kneaddata_database --download human_transcriptome bowtie2 $REFDIR
kneaddataは以下のコマンドで実行可能。
kneaddata \ --input1 ${fastq_R1} \ # fastq file, forward reads --input2 ${fastq_R2} \ # fastq file, reverse reads -db ${REFDIR} \ # データベース (上述の流れで準備) -o ${output} \ # output --output-prefix ${prefix} \ # --reorder \ # outputの配列をinputと同じ順番で並べる (default: off) -p ${process} \ # プロセス数 -t ${threads} \ # スレッド数 --max-memory 80g \ # 使用メモリ上限 --trimmomatic ${path_trimmomatic} \ # Trimmomaticの場所 (default: $PATH) --run-trim-repetitive \ # FASTQCで出てくるoverrepresented sequencesを除外するオプション (default: off) --trimmomatic-options="ILLUMINACLIP:TruSeq3-SE.fa:2:30:10" \ # Trimmomaticのオプション (オプションごとに分ける必要がある) --trimmomatic-options="SLIDINGWINDOW:4:20" \ --trimmomatic-options="MINLEN:50" \ --bowtie2 ${path_bowtie2} \ # Botwie2の場所 (default: $PATH) --bowtie2-options ${bowtie2_option} \ # default: --very-sensitive-local --decoantaminate-pairs {strict, lanient, unpaired}\ # strict: forward/reverseの片方でもマップされれば除外 (default) --run-trf # tandem repeat に対応する配列を除外 (default: on)